As he went along, he saw a man blind from birth. His disciples asked him, “Rabbi, who sinned, this man or his parents, that he was born blind?”
“Neither this man nor his parents sinned,” said Jesus, “but this happened so that the works of God might be displayed in him. As long as it is day, we must do the works of him who sent me. Night is coming, when no one can work. While I am in the world, I am the light of the world.” (John 9:1-5)
People sometimes wonder whether a modern-day physician is expected to believe in evolution. No, but there is an expectation that evolutionary concepts will be taught in medical school including the ideas surrounding genetic drift, transposons (a DNA sequence that can change its position within the genome), or small-interfering RNA to name a few. Simply put, one can be a physician and treat infections, stabilize fractures, or treat hypertension without thinking too deeply about how these diseases came into place from a biologic-genetic-evolutionary standpoint. However, as one studies various diseases, the evolutionary impact becomes quite clear, and often further exploration of the genetic history of certain diseases leads to downstream therapeutic benefit, including pharmacologic advancement.
I am a pediatric gastroenterologist, so I deal with children with intestinal, liver, and nutrition issues. Besides patient care, I also participate in medical student teaching as well as clinical research at my academic medical center. I know of many gastrointestinal diseases that have a fascinating evolutionary aspect – celiac disease, Crohn’s disease, and ulcerative colitis – as examples. In fact, my interest in the evolutionary aspect of gastroenterology began early in my career when I was involved in the description of a potential founder effect seen in the Navajo nation regarding a rare disorder known as microvillous inclusion disease. A conversation about that specific disease can occur at another time (it is quite fascinating), but my work on this project allowed me to think about multi-generational genetic mutation effects.
I want to talk about one of my clinical interests, namely cystic fibrosis (CF), its clinical consequences, and its genetics. CF occurs in about 30,000 adults and children in the United States, mainly in Caucasians although all ethnicities can have the disease. When the term “cystic fibrosis” is used, the average person will think about a severe, progressive lung disease resulting in the early death of a child. While lung disease certainly occurs in most cases, its lung presentation can be quite variable with some patients having very mild symptoms such as a mild cough and others having very severe, irreversible lung damage. Many other organs can be affected by CF, including the pancreas, liver, intestine, and gallbladder, bone, and the reproductive organs.
The cause of CF is known. Interestingly, the genetic defect was detected by Dr. Francis Collins of BioLogos and others in 1989. These scientists found a defect on chromosome 7 (the seventh chromosome) in which three nucleotide base pairs were deleted in CF patients leading to a loss of the amino acid, phenylalanine. Remember from your biology class in high school or college that DNA consists of four base pairs (adenine, thymine, guanine, and cytosine) that exist by the billions to form the genetic code of DNA. Three base pairs in a row code for one amino acid, and amino acids eventually link up to form proteins that assist in all functions of the cell. In the case of CF, these three base pairs were found to affect the formation of this one amino acid (phenylalanine) which leads to downstream disruption of specific protein function. Additionally, these same three base pairs belong to a specific gene, CFTR, or the Cystic Fibrosis Transmembrane Conductance Regulator gene.
Here is where this genetic mutation leads to fascinating, and at the same time, frankly awful consequences of CF. If both parents carry one CFTR gene mutation (heterozygous) and a child then inherits two such mutations (homozygous) from the parents, the resultant protein made from the CFTR gene malfunctions due to the single amino acid loss. The normal mechanism for the CFTR protein (remember the CFTR protein is formed from the CFTR gene) is to pump negatively-charged chloride from the cell to the outside environment while subsequently bringing positively-charged sodium back into the cell.The positive (+) and negative (-) charges of sodium and chloride, respectively, keep a neutral balance to keep water hypotonic (or at a low concentration of solutes). This water is what you would see in normal body sweat or other body secretions that are inherent for normal physiology to maintain cellular structure, reduce inflammation, and to keep harmful bacteria away from cells.
However, this mechanism changes drastically in CF. A mutation in the CFTR gene disrupts this entire function.In CF, chloride transport is impaired; hence, chloride and water moving from the cell to the outside environment become stuck inside the cell. This transport defect causes secretions to thicken. Now, the cell senses that chloride is not being secreted, and it will bring more sodium (as well as water) into the cell, further reducing the amount of water in the extra-cellular environment. This perturbed mechanism affects many of cells in the body, billions of them, leading to organ dysfunction essentially from thickened secretions as cells have an impaired release of fluid. As a result of thickened pulmonary secretions that cannot clear, bacteria can enter the lungs to cause inflammation which further decreases secretions.
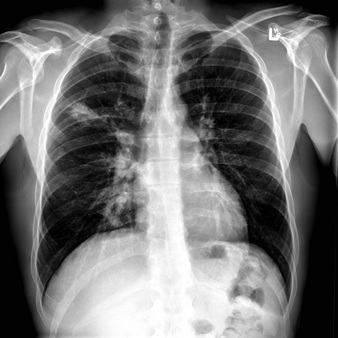
Figure 4: This chest X-ray demonstrates the classic findings of cystic fibrosis in the lungs of a child. Due to thickened lung secretions, bacteria begin to colonize lung tissue as the lung’s ability to clear bacteria is impaired. Progressive and irreversible lung disease occurs, which can ultimately lead to death over months or years.
Most people are familiar with the lung problems associated with CF.These children often are breathing quickly and may require supplemental oxygen due to poor air exchange due to thickened pulmonary secretions. However, all organs utilizing a water gradient can become diseased and progress to organ damage as a result of this mutation. Organs such as the pancreas, liver, intestine, stomach, and the reproductive organs will undergo significant injury that affects quality of life and decreases lifetime survival.
Today, there are thousands of described CFTR gene mutations although the most common is the one initially described by Dr. Collins and others—this mutation involves the abnormal coding for the amino acid, phenylalanine, at position 508 of the CFTR gene (called “delta” or ∆F508). Some mutations, such as ∆F508, cause significant disease, while others are barely noticeable so that some patients with mild CFTR gene mutations may not be diagnosed with CF for many decades. The growing knowledge of these genetic mutations and their downstream organ effects have allowed CF medical teams to better determine treatment options and prolong lifespans while also improving the quality of life of these patients.
One would think that these mutations are pointless, cause needless suffering, and have nothing but chronic, tragic consequences. However, we need to evaluate this mutation again—remember that a parent of a CF child typically will have one CFTR gene mutation, not two mutations as in the affected child. Would a heterozygote mutation have an evolutionary advantage?
It turns out the answer is “Yes…probably” (remember that good science is always full of qualifiers). Genomic research has shown that the mutation for ∆F508 probably started as a one-time occurrence 58,000 to 173,000 years ago. ∆F508 mutations may have protected against infections in Paleolithic Europe, including Salmonella typhi (typhoid) and influenzae. One infection, Vibrio cholerae (cholera) may have, in particular, been influenced by this heterozygous inheritance. Cholera infection occurs through drinking a contaminated water supply, leading quickly to profound, massive, watery diarrhea with resultant dehydration, shock, and death. However, research has suggested that CFTR protein mutations may cause V. cholerae to have difficulty adhering to the lining of the small intestine while causing less fluid to be secreted during an active infection. It would make sense that a mutation of the CFTR protein would be protective in situations where there was a contaminated water supply, poor hygiene, and a poor understanding of disease transmission.
There also appears to be an advantage to having a CFTR protein mutation in tuberculosis prevention. Epidemiologic evidence from British mortality records in the 1800s followed by modeling has suggested that CFTR protein mutations protect from tuberculosis (the reason is not entirely clear), and that the number of patients with cystic fibrosis may be declining in those parts of the world where tuberculosis becomes an irrelevant pathogen. These evolutionary aspects are fascinating and provocative, but we have to be very careful to make sure we are not making the genetics history of CF a “just so story” (or an ad hoc fallacy).
Theological Reflections
Now that we have gone into quite a bit of detail about one specific disease, I want to describe certain aspects of a Creator related to what we have learned about CF genetics.
God is smarter than me (or you). It is amazing what He has done in regards to the complexity of life and inheritance.
Disease and death are a part of reality, which is justified in a reading of the Bible. The blind man, just like a child with CF, had no sin which caused a debilitating illness. Much of the suffering that we see in the world of medicine is due to environmental and genetic consequences.
Besides our immune system, there appear to be other mechanisms for protection against disease that are often difficult to observe in a first-world setting. We see this idea in how CFTR protein mutations may protect against certain infectious diseases. Whether God did this through a divine purpose or allowed a random mechanism to occur (or for what appears to be random to us, in our limited scientific understanding) is irrelevant. I can see that the process of mutation occurs, and I believe in a God that allows it.
Finally, I am thankful that “modern” medicine has developed therapies in the basic science and clinical realms to allow an improved quality of life in CF patients, as well as for patients with other diseases. It is now common for patients with ∆F508 mutations to live into their 30s or 40s. At the beginning of the 20th century, most CF children died well before two years of age. I use quotation marks around this word – modern – as 20 years from today, much of our current CF treatments will have changed, leading to even longer life for these patients. I am thankful that God has allowed us the capacity to investigate our world at an ever-advancing state through the use of the scientific method. That, in itself, is a gift for humanity for which we should always be thankful.